
Raji Chem World Search Engine

Custom Search
Wednesday, August 31, 2011

Alternative to Benzoquinone for Room-Temperature Fujiwara–Moritani Reactions

tert-Butyl perbenzoate is a substitute for benzoquinone for mild (room-temperature) Fujiwara–Moritani reactions between acetanilides and butyl acrylate under homogeneous conditions. The system was enhanced further by including Cu(OAc)2 as a cocatalyst. Methyl methacrylate can be activated toward coupling under these conditions.
Synthesis of Cyclic Enones via Direct Palladium-Catalyzed Aerobic Dehydrogenation of Ketones

α,β-Unsaturated carbonyl compounds are versatile intermediates in the synthesis of pharmaceuticals and biologically active compounds. Here, we report the discovery and application of Pd(DMSO)2(TFA)2 as a catalyst for direct dehydrogenation of cyclohexanones and other cyclic ketones to the corresponding enones, using O2 as the oxidant. The substrate scope includes heterocyclic ketones and several natural-product precursors.
Sequential Catalysis for the Production of Sterically Hindered Amines: Ru(II)-Catalyzed C–H Bond Activation and Hydrosilylation of Imines

Sequential ruthenium(II) catalyzed reactions for the production of secondary amines from simple imines are reported. They involve Ru(II)-acetate based catalytic systems for C–H Bond activation/diarylation of simple imines with aryl bromides followed by [RuCl2(arene)]2hydrosilylation of diarylated imines. The direct diarylation of aldimines can be profitably produced by Ru(II) catalytic systems in the presence of both acetate and PPh3 ligands. By contrast, PPh3 additive disfavors diarylation of ketimines that is achieved with [Ru(OAc)2(p-cymene)] alone. The catalytic hydrosilylation of the resulting imines was simply performed with H2SiPh2 in the presence of [RuCl2(arene)]2 catalyst at room temperature leading to overall yield of 70–86% of secondary amines
ACS Catalysis, 2011, 1257.
ACS Catalysis, 2011, 1257.
Tuesday, August 30, 2011
Selective, Nickel-Catalyzed Hydrogenolysis of Aryl Ethers
Selective hydrogenolysis of the aromatic carbon-oxygen (C-O) bonds in aryl ethers is an unsolved synthetic problem important for the generation of fuels and chemical feedstocks from biomass and for the liquefaction of coal. Currently, the hydrogenolysis of aromatic C-O bonds requires heterogeneous catalysts that operate at high temperature and pressure and lead to a mixture of products from competing hydrogenolysis of aliphatic C-O bonds and hydrogenation of the arene. Here, we report hydrogenolyses of aromatic C-O bonds in alkyl aryl and diaryl ethers that form exclusively arenes and alcohols. This process is catalyzed by a soluble nickel carbene complex under just 1 bar of hydrogen at temperatures of 80 to 120°C; the relative reactivity of ether substrates scale as Ar-OAr>>Ar-OMe>ArCH2-OMe (Ar, Aryl; Me, Methyl). Hydrogenolysis of lignin model compounds highlights the potential of this approach for the conversion of refractory aryl ether biopolymers to hydrocarbons.

Science 22 April 2011:
Vol. 332 no. 6028 pp. 439-443 ,DOI: 10.1126/science.1200437
Proton-Catalyzed, Silane-Fueled Friedel-Crafts Coupling of Fluoroarenes

Science 29 April 2011:
Vol. 332 no. 6029 pp. 574-577
DOI: 10.1126/science.1202432
Vol. 332 no. 6029 pp. 574-577
DOI: 10.1126/science.1202432
Palladium-Catalyzed Aerobic Dehydrogenation of Substituted Cyclohexanones to Phenols

Aromatic molecules are key constituents of many pharmaceuticals, electronic materials, and commodity plastics. The utility of these molecules directly reflects the identity and pattern of substituents on the aromatic ring. Here, we report a palladium(II) catalyst system, incorporating an unconventional ortho-dimethylaminopyridine ligand, for the conversion of substituted cyclohexanones to the corresponding phenols. The reaction proceeds via successive dehydrogenation of two saturated carbon-carbon bonds of the six-membered ring and uses molecular oxygen as the hydrogen acceptor. This reactivity demonstrates a versatile and efficient strategy for the synthesis of substituted aromatic molecules with fundamentally different selectivity constraints from the numerous known synthetic methods that rely on substitution of a preexisting aromatic ring.
Science 8 July 2011:
Vol. 333 no. 6039 pp. 209-213
DOI: 10.1126/science.1204183
Vol. 333 no. 6039 pp. 209-213
DOI: 10.1126/science.1204183
Catalysis for fluorination and trifluoromethylation

Recent advances in catalysis have made the incorporation of fluorine into complex organic molecules easier than ever before, but selective, general and practical fluorination reactions remain sought after. Fluorination of molecules often imparts desirable properties, such as metabolic and thermal stability, and fluorinated molecules are therefore frequently used as pharmaceuticals or materials. But the formation of carbon−fluorine bonds in complex molecules is a significant challenge. Here we discuss reactions to make organofluorides that have emerged within the past few years and which exemplify how to overcome some of the intricate challenges associated with fluorination.

Copper-Catalyzed Trifluoromethylation of Unactivated Olefins - Parsons - 2011 - Angewandte Chemie International Edition - Wiley Online Library

Activating the inactive: A copper-catalyzed allylic trifluoromethylation of unactivated terminal olefins proceeds under mild conditions to produce linear allylic trifluoromethylated products with high E/Z selectivity (see scheme). The reaction can be applied to a range of substrates bearing numerous functional groups. Furthermore, the reaction is scalable and amenable to a benchtop setup.
DOI: 10.1002/anie.201104053
Remarkably High Reactivity of Pd(OAc)2/Pyridine Catalysts: Nondirected C[BOND]H Oxygenation of Arenes -

Less is more: The rational optimization and general applicability of the catalytic system Pd(OAc)2/pyridine is described (see scheme). The catalyst shows excellent reactivity in the C![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H oxygenation of simple aromatic substrates. The Pd/pyridine ratio is critical as the use of 1 equivalent of pyridine per Pd center leads to dramatic enhancements in both reactivity and site selectivity in comparison to Pd(OAc)2alone.
H oxygenation of simple aromatic substrates. The Pd/pyridine ratio is critical as the use of 1 equivalent of pyridine per Pd center leads to dramatic enhancements in both reactivity and site selectivity in comparison to Pd(OAc)2alone.
H oxygenation of simple aromatic substrates. The Pd/pyridine ratio is critical as the use of 1 equivalent of pyridine per Pd center leads to dramatic enhancements in both reactivity and site selectivity in comparison to Pd(OAc)2alone.
M.Sanford et al. DOI: 10.1002/anie.201103327
Monday, August 29, 2011
2011-GATE CHEMISTRY QUESTION PAPER AND ANSWER DOWNLODE
2011-GATE CHEMISTRY QUESTION PAPER
2011-GATE CHEMISTRY ANSWER KEY
For more previous year question,
Click Here
2011-GATE CHEMISTRY ANSWER KEY
For more previous year question,
Click Here
Saturday, August 27, 2011



Cobalt-Catalyzed Chemoselective Insertion of Alkene into the Ortho C−H Bond of Benzamide - Journal of the American Chemical Society (ACS Publications)

Insertion of 1-alkene, 2-alkene, and styrene into the ortho C−H bond of benzamide in the presence of an inexpensive cobalt catalyst, DMPU as a crucial ligand, and cyclohexylmagnesium chloride proceeds smoothly at 25 °C to selectively give the ortho-alkylated product. Notable features of this reaction include the structural variety of the alkene and the amide substrate and the tolerance of functional groups such as halide, olefin, ester, and amide groups.
Nickel-catalyzed direct C–H arylation of unactivated arenes with aryl halides
Readily available Ni(OAc)2·4H2O could catalyze direct C–H arylation of unactivated arenes with aryl bromides/iodides in presence of 1,10-phenanthroline as the ligand without using any additives. This protocol makes C–H arylation more practical and useful.
Intramolecular Oxidative C−N Bond Formation
Intramolecular Oxidative C−N Bond Formation for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions

New synthetic procedures for intramolecular oxidative C−N bond formation have been developed for the preparation of carbazoles starting from N-substituted amidobiphenyls under either Cu-catalyzed or metal-free conditions using hypervalent iodine(III) as an oxidant. Whereas iodobenzene diacetate or bis(trifluoroacetoxy)iodobenzene alone undergoes the reaction to provide carbazole products in moderate to low yields, combined use of copper(II) triflate and the iodine(III) species significantly improves the reaction efficiency, giving a more diverse range of products in good to excellent yields. On the basis of mechanistic studies including kinetic profile, isotope effects, and radical inhibition experiments, the copper species is proposed to catalytically activate the hypervalent iodine(III) oxidants. The synthetic utility of the present approach was nicely demonstrated in a direct synthesis of indolo[3,2-b]carbazole utilizing a double C−N bond formation.

Palladium-Catalyzed Oxidative sp2 C−H Bond Acylation with Alcohols

An efficient method was developed for the direct acylation of arene sp2 C−H bonds with alcohols using palladium chloride as a catalyst and peroxide as the oxidant. The alcohols were oxidized to their corresponding aldehydes in situ and efficiently coupled with 2-arylpyridines to give aryl ketones in chlorobenzene.
Org. Lett., 2011, 13 (7), pp 1614–1617
Org. Lett., 2011, 13 (7), pp 1614–1617
Palladium-Catalyzed ortho-Acylation of Acetanilides with Aldehydes through Direct C[BOND]H Bond Activation

Easy access too-acyl acetanilides: A new Pd-catalyzed ortho-acylation of acetanilides with both aromatic and aliphatic aldehydes has been developed based on a C
H activation process. In the presence of tert-butyl hydroperoxide (TBHP) as the ideal oxidant, this reaction provides an efficient access to ortho-acyl acetanilides in good yields (see scheme).doi/10.1002/chem.201101192
Thursday, August 25, 2011
C–H Bond Functionalization of Benzoic Acid: Catalytic Synthesis of 2-Hydroxy-6H-benzo[c]chromen-6-ones Using (Cp*IrCl2)2

Catalytic H/D exchange reactions of benzene and benzoic acid with deuterated solvents have been studied using (Cp*IrCl2)2. A 1:1 mixture of D2O/CD3OD produced the highest turnover numbers for benzene. High levels of deuterium incorporation into benzoic acid were observed only when sodium acetate was added to the reaction mixture. Attempts at producing hydroxybenzoic acid by catalytic C–H functionalization of benzoic acid with benzoquinone were unsuccessful. Instead, 2-hydroxy-6H-benzo[c]chromen-6-one was isolated as the major product. An array of substituted benzoic acids was analyzed for this functionalization reaction. Preliminary mechanistic studies indicate that the benzochromenones are formed by C–H bond activation of benzoic acid followed by insertion of benzoquinone into the iridium–carbon bond.
Palladium-Catalyzed Alkylation of ortho-C(sp2)–H Bonds of Benzylamide Substrates with Alkyl Halides

A highly efficient and generally applicable method has been developed to functionalize theortho-C(sp2)–H bonds of picolinamide (PA)-protected benzylamine substrates with a broad range of β-H-containing alkyl halides. Sodium triflate has been identified as a critical promoter for this reaction system. The PA group can be easily installed and removed under mild conditions. This method provides a new strategy to prepare highly functionalized benzylamines for the synthesis of complex molecules.
Palladium(II)-Catalyzed Direct Alkoxylation of Arenes: Evidence for Solvent-Assisted Concerted Metalation Deprotonation

Density functional theory investigations on the mechanism of palladium acetate catalyzed direct alkoxylation of N-methoxybenzamide in methanol reveal that the key steps involve solvent-assisted N–H as well as C–H bond activations. The transition state for the critical palladium–carbon bond formation through a concerted metalation deprotonation (CMD) process leading to a palladacycle intermediate has been found to be more stable in the methanol-assisted pathway as compared to an unassisted route.

Synthesis of Quinolines via Rh(III)-Catalyzed Oxidative Annulation of Pyridines

Selective synthesis of quinolines has been achieved via oxidative annulation of functionalized pyridines with two alkyne molecules under Rh(III)-catalyzed cascade C–H activation of pyridines using Cu(OAc)2 as an oxidant. The selectivity of this reaction is oxidant-dependent, particularly on the anion of the oxidant.
DOI: 10.1021/jo201266u
DOI: 10.1021/jo201266u
Palladium-Catalyzed Synthesis of Dibenzophosphole Oxides via Intramolecular Dehydrogenative Cyclization

Dibenzophosphole oxides were obtained from secondary hydrophosphine oxides with a biphenyl group by dehydrogenation via phosphine–hydrogen and carbon–hydrogen bond cleavage in the presence of a catalytic amount of palladium(II) acetate, Pd(OAc)2. By using this reaction, a ladder-type dibenzophosphole oxide could also be synthesized by double intramolecular dehydrogenative cyclization.
Wednesday, August 24, 2011
Neocuproine–KOtBu promoted intramolecular cross coupling to approach fused rings
Polycycles can be produced with different linkages (A, B = O, N, C, S) by constructing biaryl C–C bonds via neocuproine–KOtBu promoted cross coupling between C–Xs and C–Hs.

Friday, August 19, 2011
Towards mild metal-catalyzed C–H bond activation
Functionalizing traditionally inert carbon–hydrogen bonds represents a powerful transformation in organic synthesis, providing new entries to valuable structural motifs and improving the overall synthetic efficiency. C–H bond activation, however, often necessitates harsh reaction conditions that result in functional group incompatibilities and limited substrate scope. An understanding of the reaction mechanism and rational design of experimental conditions have led to significant improvement in both selectivity and applicability. This critical reviewsummarizes and discusses endeavours towards the development of mild C–H activation methods and wishes to trigger more research towards this goal. In addition, we examine select examples in complex natural product synthesis to demonstrate the synthetic utility of mild C–H functionalization (84 references).

Wednesday, August 17, 2011
A General Method for Copper-Catalyzed Arene Cross-Dimerization
A general method for a highly regioselective copper-catalyzed cross-coupling of two aromatic compounds using iodine as an oxidant has been developed. The reactions involve an initial iodination of one arene followed by arylation of the most acidic C–H bond of the other coupling component. Cross-coupling of electron-rich arenes, electron-poor arenes, and five- and six-membered heterocycles is possible in many combinations. Typically, a 1/1.5 to 1/3 ratio of coupling components is used, in contrast to existing methodology that often employs a large excess of one of the arenes. Common functionalities such as ester, ketone, aldehyde, ether, nitrile, nitro, and amine are well-tolerated.

Pd(II)-Catalyzed para-Selective C–H Arylation of Monosubstituted Arenes

Pd-catalyzed highly para-selective C–H arylation of monosubstituted arenes (including toluene) is developed for the first time using an F+ reagent as a bystanding oxidant. This finding provides a new retrosynthetic disconnection for para-substituted biaryl synthesis via C–H/C–H cross-coupling.
Enantioselective α-Arylation of Carbonyls via Cu(I)-Bisoxazoline Catalysis
The enantioselective α-arylation of both lactones and acyl oxazolidones has been accomplished using a combination of diaryliodonium salts and copper catalysis. These mild catalytic conditions provide a new strategy for the enantioselective construction and retention of enolizable α-carbonyl benzylic stereocenters, a valuable synthon for the production of medicinal agents.

Enantioselective α-Arylation of N-Acyloxazolidinones with Copper(II)-bisoxazoline Catalysts and Diaryliodonium Salts
A new strategy for the catalytic enantioselective α-arylation of N-acyloxazolidinones with chiral copper(II)-bisoxazoline complexes and diaryliodonium salts is described. The mild catalytic conditions are operationally simple, produce valuable synthetic building blocks in excellent yields and enantioselectivities, and can be applied to the synthesis of important nonsteroidal anti-inflammatory agents and their analogues.

Iron-catalyzed aryl- and alkenyllithiation of alkynes and its application to benzosilole synthesis
Phenyl- and vinyllithiums having an alkyl substituent at their ortho- and cis-position, respectively, readily added to alkynes in the presence of 5 mol% of Fe(acac)3. The reaction of o-(trimethylsilyl)phenyllithium with alkynes gave benzosiloles through an addition–cyclization sequence.
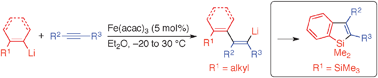
Friday, August 12, 2011
Expedient Synthesis of N-Acyl Anthranilamides and β-Enamine Amides by the Rh(III)-Catalyzed Amidation of Aryl and Vinyl C–H Bonds with Isocyanates

A Rh(III)-catalyzed protocol for the amidation of anilide and enamide C–H bonds with isocyanates has been developed. This method provides direct and efficient syntheses of N-acyl anthranilamides, enamine amides, and pyrimidin-4-one heterocycles.
Tuesday, August 9, 2011
Palladium-Catalyzed ortho-Acylation of Acetanilides with Aldehydes through Direct C[BOND]H Bond Activation
Easy access too-acyl acetanilides: A new Pd-catalyzed ortho-acylation of acetanilides with both aromatic and aliphatic aldehydes has been developed based on a CH activation process. In the presence of tert-butyl hydroperoxide (TBHP) as the ideal oxidant, this reaction provides an efficient access to ortho-acyl acetanilides in good yields (see scheme).
Chem. Eur. J, 2011, DOI: 10.1002/chem.201101192
H activation process. In the presence of tert-butyl hydroperoxide (TBHP) as the ideal oxidant, this reaction provides an efficient access to ortho-acyl acetanilides in good yields (see scheme).Chem. Eur. J, 2011, DOI: 10.1002/chem.201101192
Sunday, August 7, 2011
Synthesis of 1-Aminoisoquinolines via Rh(III)-Catalyzed Oxidative Coupling

[RhCp*Cl2]2 can catalyze the oxidative coupling of N-aryl and N-alkyl benzamidines with alkynes to give N-substituted 1-aminoisoquinolines in high selectivity.
Palladium-Catalyzed Intramolecular C(sp2)–H Amidination by Isonitrile Insertion Provides Direct Access to 4-Aminoquinazolines from N-Arylamidines

An efficient method for the synthesis of 4-amino-2-aryl(alkyl)quinazolines from readily available N-arylamidines and isonitriles via palladium-catalyzed intramolecular aryl C–H amidination by isonitrile insertion has been developed.
Palladium-Catalyzed Direct 2-Alkylation of Indoles by Norbornene-Mediated Regioselective Cascade C–H Activation

A palladium-catalyzed direct 2-alkylation reaction of free N-H indoles has been developed. This reaction relies on a norbornene-mediated cascade C–H activation process at the indole ring, which features high regioselectivity and excellent functional group tolerance. The reaction represents the first example for a generally applicable, direct C–H alkylation of indole at the 2-position
Palladium-Catalyzed Direct Ethynylation of C(sp3)–H Bonds in Aliphatic Carboxylic Acid Derivatives

The first catalytic alkynylation of unactivated C(sp3)–H bonds has been accomplished. The method allows for the straightforward introduction of an ethynyl group into aliphatic acid derivatives under palladium catalysis. This new reaction can be applied to the rapid elaboration of complex aliphatic acids, for example, via azide/alkyne cycloaddition.
Naoto Chatani, JACS, 2011, DOI: 10.1021/ja206002m
Naoto Chatani, JACS, 2011, DOI: 10.1021/ja206002m
Saturday, August 6, 2011
DMF Distillation
The Protocol of DMF Distillation
1. First add DMF (from the carboy) to the filteration flask(2L),and then add two spoons of Calcium Hydride.
2. Stir DMF for around 4hours (with vigorous stirring) or over night. (To get rid of water)
3. Stop the stirring and let DMF bottle stand for a while. And then filter DMF to remove the base. (The calcium hydride can be reused for next time, you could add crude DMF into the flask directly)
4. Add the filtrated DMF into the big RBF of distillation apparatus inside the solvent room. Turn on the circulation water, heating mantle and the pump.
5. Reflux DMF for 1h without collecting (remove the amine) After that start collecting the DMF.
Attention:
a. In the first step, we can’t change the sequence of adding DMF fisrt and then Calcium Hydride,which will absorb the water in the air.
Tuesday, August 2, 2011
Palladium-Catalyzed Direct Cross-Coupling Reaction of Glycals with Activated Alkenes
An efficient method for a Pd(OAc)2-catalyzed cross-coupling reaction of glycals with activated alkenes under mild conditions has been developed. This transformation provides an expedient synthetic method to C(2)-functionalized glycals, which are common structural building blocks in natural products and other biologically active compounds. The reaction scope includes different kinds of carbohydrates, protecting groups and substituents on alkene. Moderate to excellent yields and pure E configuration selectivity were obtained.

Rhodium-Catalyzed Oxidative ortho-Acylation of Benzamides with Aldehydes: Direct Functionalization of the sp2 C–H Bond
A rhodium-catalyzed oxidative acylation of benzamides with aryl aldehydes via direct sp2 C–H bond cleavage is described. In the presence of [Cp*RhCl2]2, AgSbF6, and silver carbonate as an oxidant, N,N-diethyl benzamides can be effectively carbonylated to yield ortho-acyl benzamides.

Palladium(II)-Catalyzed Direct Intermolecular Alkenylation of Chromones
A new efficient method for the direct alkenylation of chromones via a palladium(II)-catalyzed C–H functionalization reaction was developed. The use of pivalic acid with Cu(OAc)3/Ag2CO3provided superior reactivity in the cross-coupling of chromones with alkene partners. This approach represents a significant advance over the existing two-step method and afforded various 3-vinylchromone derivatives, which are privileged structures in many biologically active compounds and versatile synthetic building blocks.

A Catalyst-Free Benzylic C–H Bond Olefination of Azaarenes for Direct Mannich-like Reactions
A highly efficient synthesis of trans-alkenylazaarene under catalyst-free conditions was developed via the addition of methylazaarenes to N-sulfonyl aldimines and a subsequent C–N elimination in situ. A one-pot procedure for this addition–elimination was also developed. The reaction could tolerate a broad substrate scope and give the corresponding alkenylazaarenes in high yields.

Direct Transformation of N,N-Dimethylformamide to −CN: Pd-Catalyzed Cyanation of Heteroarenes via C–H Functionalization
This paper describes the direct cyanation of indoles and benzofurans employing N,N-dimethylformamide (DMF) as both reagent and solvent. Isotopic labeling experiments indicated that both the N and the C of the cyano group derived from DMF. This transformation offers an alternative method for preparing aryl nitriles, though it is currently limited in scope to indoles and benzofurans.

Monday, August 1, 2011
Organocatalytic, Oxidative, Intramolecular CH Bond Amination and Metal-free Cross-Amination of Unactivated Arenes at Ambient Temperature
The twinkling of an I: In a new atom-economical and environmentally friendly organocatalytic method for intramolecular CH amination, the CN bond forms at ambient temperature by abstraction of two atoms of hydrogen; only acetic acid and water are formed as by-products. The method has also been extended to the unprecedented metal-free cross-amination of nonactivated arenes.

H amination, the CN bond forms at ambient temperature by abstraction of two atoms of hydrogen; only acetic acid and water are formed as by-products. The method has also been extended to the unprecedented metal-free cross-amination of nonactivated arenes.
Subscribe to:
Comments (Atom)