
Raji Chem World Search Engine

Custom Search
Tuesday, January 31, 2012






Iron-Catalyzed Regio- and Stereoselective Chlorosulfonylation of Terminal Alkynes with Aromatic Sulfonyl Chlorides

Terminal alkynes react with aromatic sulfonyl chlorides in the presence of an iron(II) catalyst and a phosphine ligand to give (E)-β-chlorovinylsulfones with 100% regio- and stereoselectivity. Various functional groups, such as chloride, bromide, iodide, nitro, ketone, and aldehyde, are tolerated under the reaction conditions. Addition of tosyl chloride to a 1,6-enyne followed by radical 5-exo-trig cyclization gave an exocyclic alkenylsulfone.

Cu-Catalyzed Oxidative C(sp2)–H Cycloetherification of o-Arylphenols for the Preparation of Dibenzofurans

A new process involving copper-catalyzed aerobic C(sp2)–H activation, followed by cycloetherification, has been developed. This reaction serves as a direct method for the preparation of multisubstituted dibenzofurans starting with o-arylphenols. The presence of a strong para-electron-withdrawing group (e.g., NO2) on the phenol is essential for the success of the reaction.




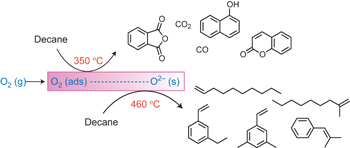
Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C–H Oxidation Reactions Catalyzed by Pd (and Cu)

Oxidation reactions are key transformations in organic chemistry because they can increase chemical complexity and incorporate heteroatom substituents into carbon-based molecules. This principle is manifested in the conversion of petrochemical feedstocks into commodity chemicals and in the synthesis of fine chemicals, pharmaceuticals, and other complex organic molecules. The utility and function of these molecules correlate directly with the presence and specific placement of oxygen and nitrogen heteroatoms and other functional groups within the molecules.
Methods for selective oxidation of C–H bonds have expanded significantly over the past decade, and their role in the synthesis of organic chemicals will continue to increase. Our group’s contributions to this field are linked to our broader interest in the development and mechanistic understanding of aerobic oxidation reactions. Molecular oxygen (O2) is the ideal oxidant. Its low cost and lack of toxic byproducts make it a highly appealing reagent that can address key “green chemistry” priorities in industry. With strong economic and environmental incentives to use O2, the commmodity chemicals industry often uses aerobic oxidation reactions. In contrast, O2 is seldom used to prepare more-complex smaller-volume chemicals, a limitation that reflects, in part, the limited synthetic scope and utility of existing aerobic reactions.
Pd-catalyzed reactions represent some of the most versatile methods for selective C–H oxidation, but they often require stoichiometric transition-metal or organic oxidants, such as CuII, AgI, or benzoquinone. This Account describes recent strategies that we have identified to use O2 as the oxidant in these reactions. In Pd-catalyzed C–H oxidation reactions that form carbon-heteroatom bonds, the stoichiometric oxidant is often needed to promote difficult reductive elimination steps in the catalytic mechanism. To address this challenge, we have identified new ancillary ligands for Pd that promote reductive elimination, or replaced Pd with a Cu catalyst that undergoes facile reductive elimination from a CuIII intermediate. Both strategies have enabled O2 to be used as the sole stoichiometric oxidant in the catalytic reactions. C–H oxidation reactions that form the product via β-hydride or C–C reductive elimination steps tend to be more amenable to the use of O2. The use of new ancillary ligands has also overcome some of the limitations in these methods. Mechanistic studies are providing insights into some (but not yet all) of these advances in catalytic reactivity.
Iron-Catalyzed Intramolecular Allylic C–H Amination

A highly selective C–H amination reaction under iron catalysis has been developed. This novel system, which employs an inexpensive, nontoxic [FeIIIPc] catalyst (typically used as an industrial ink additive), displays a strong preference for allylic C–H amination over aziridination and all other C–H bond types (i.e., allylic > benzylic > ethereal > 3° > 2° 1°). Moreover, in polyolefinic substrates, the site selectivity can be controlled by the electronic and steric character of the allylic C–H bond. Although this reaction is shown to proceed via a stepwise mechanism, the stereoretentive nature of C–H amination for 3° aliphatic C–H bonds suggests a very rapid radical rebound step.
Intermolecular Ritter-Type C–H Amination of Unactivated sp3 Carbons

Intermolecular Ritter-type C–H amination of unactivated sp3 carbons has been developed. This new reaction proceeds under mild conditions using readily available reagents and an inexpensive source of nitrogen (acetonitrile). A broad scope of substrates can be aminated with this method since many functional groups are tolerated. This reaction also allows for the direct, innate C–H amination of a variety of hydrocarbons such as cyclohexane without the need of prefunctionalization or installation of a directing group.
Copper-Mediated Sequential Cyanation of Aryl C–B and Arene C–H Bonds Using Ammonium Iodide and DMF

The cyanation of aromatic boronic acids, boronate esters, and borate salts was developed under copper-mediated oxidative conditions using ammonium iodide and DMF as the source of nitrogen and carbon atom of the cyano unit, respectively. The procedure was successfully extended to the cyanation of electron-rich benzenes, and regioselective introduction of a cyano group at the arene C–H bonds was also achieved. The observation that the reaction proceeds via a two-step process, initial iodination and then cyanation, led us to propose that ammonium iodide plays a dual role to provide iodide and nitrogen atom of the cyano moiety.
Zinc(II)-Catalyzed Redox Cross-Dehydrogenative Coupling of Propargylic Amines and Terminal Alkynes for Synthesis of N-Tethered 1,6-Enynes

The zinc(II)-catalyzed redox cross-dehydrogenative coupling (CDC) of propargylic amines and terminal alkynes proceeds to afford N-tethered 1,6-enynes. In the current CDC reaction, a C(sp)–C(sp3) bond is formed between the carbon adjacent to the nitrogen atom in the propargylic amine and the terminal carbon of the alkyne with reduction of the C–C triple bond of the propargylic amine, which acts as an internal oxidant.
Mirror symmetry breaking and chiral amplification in foldamer-based supramolecular helical aggregates

Spontaneous asymmetric generation of supramolecular chiral fibers was observed in the folding induced self-assembly of a lock-washer shaped foldamer. A secondary nucleation growth mechanism is proposed to explain the observed chiral amplification or deracemization of these supramolecular fibers.

Saturday, January 28, 2012




Transition-metal-free Coupling Reactions of Aryl Halides
Transition metal catalysis plays a leading role in C(sp2)–C(sp2) bond formation through substitution reactions. On the other hand, a similar type of substitution reaction has recently been achieved without the aid of transition-metal catalysts. In this review, recent transition-metal-free coupling reactions of aryl halides with arenes, alkenes, or aryl Grignard reagents are summarized in view of SRN1 reaction.
Friday, January 27, 2012
Wednesday, January 25, 2012
Up the Hill: Selective Double-Bond Isomerization of Terminal 1,3-Dienes towards Z-1,3-Dienes or 2Z,4E-Dienes

Against all odds: Two different cobalt catalyst systems led to the selective isomerization of 1,3-dienes. In the case of the [CoBr2(py-imine)]-catalyzed reaction, the Z-1,3-diene was formed in a highly selective manner (see scheme). When the catalyst precursor [CoBr2(dpppMe2)] was applied, a double-bond migration and selective isomerization towards the 2Z,4E-configured 2,4-dienes were observed.

A Highly Efficient Gold-Catalyzed Oxidative C[BOND]C Coupling from C[BOND]H Bonds Using Air as Oxidant

A breath of fresh air: The title reaction has been developed for the coupling of amines with nitroalkanes and different unmodified ketones using air as the sole oxidant under mild reaction conditions. The safe, convenient, and environmentally benign process, as well as the low catalyst loading, short reaction time, and good yields make this protocol very practical (see scheme).
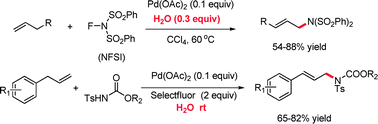
Highly Regioselective Copper-Catalyzed Benzylic C[BOND]H Amination by N-Fluorobenzenesulfonimide

Primary target: A practical and effective copper-catalyzed amination strategy for synthesizing various benzylic amines from benzylic hydrocarbons is described (see scheme; DCE=1,2-dichloroethane). Xylene substrates can undergo diamination reactions using this method. The remarkable preference for primary over secondary benzylic CH bonds has been observed for the first time.
Tuesday, January 24, 2012









Friday, January 20, 2012

A method for the synthesis of 2-aminobenzoxazoles

Abstract
A synthesis of 2-aminobenzoxazoles from the parent C–H compound is described. The procedure involves deprotonation at the 2-position of the benzoxazole and quenching the intermediate organolithium species with a halogen electrophile. The 2-halobenzoxazole is then treated in the same pot with an amine nucleophile to afford the desired product. The substrate scope and selectivity of the reaction are presented. The method is operationally simple and provides access to a variety of amine products bearing additional nucleophilic heteroatoms.
A Unique Class of Near-Infrared Functional Fluorescent Dyes with Carboxylic-Acid-Modulated Fluorescence ON/OFF Switching: Rational Design, Synthesis, Optical Properties, Theoretical Calculations, and Applications for Fluorescence Imaging in Living Animals

Fluorescence imaging is one of the most powerful techniques for monitoring biomolecules in living systems. Fluorescent sensors with absorption and emission in the near-infrared (NIR) region are favorable for biological imaging applications in living animals, as NIR light leads to minimum photodamage, deep tissue penetration, and minimum background autofluorescence interference. Herein, we have introduced a new strategy to design NIR functional dyes with the carboxylic-acid-controlled fluorescence on–off switching mechanism by the spirocyclization. Based on the design strategy, we have developed a series of Changsha (CS1–6) NIR fluorophores, a unique new class of NIR functional fluorescent dyes, bearing excellent photophysical properties including large absorption extinction coefficients, high fluorescence quantum yields, high brightness, good photostability, and sufficient chemical stability. Significantly, the new CS1–6 NIR dyes are superior to the traditional rhodamine dyes with both absorption and emission in the NIR region while retaining the rhodamine-like fluorescence ON-OFF switching mechanism. In addition, we have performed quantum chemical calculations with the B3LYP exchange functional employing 6-31G* basis sets to shed light on the structure-optical properties of the new CS1–6 NIR dyes. Furthermore, using CS2 as a platform, we further constructed the novel NIR fluorescent TURN-ON sensor 7, which is capable of imaging endogenously produced HClO in the living animals, demonstrating the value of our new CS NIR functional fluorescent dyes. We expect that the design strategy may be extended for development of a wide variety of NIR functional dyes with a suitable fluorescence-controlled mechanism for many useful applications in biological studies.
Thursday, January 19, 2012
![Graphical abstract: Synthesis of a [2.2]paracyclophane based planar chiral palladacycle by a highly selective kinetic resolution/C–H activation reaction](http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C2CC16864B)
Wednesday, January 18, 2012

Tuesday, January 17, 2012



Sunday, January 15, 2012
Enantioselective construction of quaternary N-heterocycles by palladium-catalysed decarboxylative allylic alkylation of lactams
The enantioselective synthesis of nitrogen-containing heterocycles (N-heterocycles) represents a substantial chemical research effort and resonates across numerous disciplines, including the total synthesis of natural products and medicinal chemistry. In this Article, we describe the highly enantioselective palladium-catalysed decarboxylative allylic alkylation of readily available lactams to form 3,3-disubstituted pyrrolidinones, piperidinones, caprolactams and structurally related lactams. Given the prevalence of quaternary N-heterocycles in biologically active alkaloids and pharmaceutical agents, we envisage that our method will provide a synthetic entry into the de novo asymmetric synthesis of such structures. As an entry for these investigations we demonstrate how the described catalysis affords enantiopure quaternary lactams that intercept synthetic intermediates previously used in the synthesis of the Aspidosperma alkaloids quebrachamine and rhazinilam, but that were previously only available by chiral auxiliary approaches or as racemic mixtures.

Nature Chemistry (2011) doi:10.1038/nchem.1222
Subscribe to:
Comments (Atom)